英文原题:Accurate descriptions of molecule-surface interactions in electrocatalytic CO2 reduction on the copper surfaces
铜是一些重要工业催化剂的主要成分,如水汽变换反应和合成气合成甲醇反应。此外,铜还被广泛用于催化CO2电还原成高价值的燃料或者化学品。理论模拟在这些重要催化剂的发展和理性设计中,扮演着越来越重要的角色。而与此严重不符的是,广泛应用的广义梯度近似泛函GGA的精度较低。因此,利用本课题组自主发展的组合方法XO-PBC和双杂化泛函XYG3,我们提出了一种适用于金属表面催化体系的组合方法XYG3:GGA,并成功应用于币族催化体系,实现了精确高效的分子-表面相互作用能计算。我们通过一系列吸附能和表面反应能垒的实验值验证了XYG3:GGA的精度,然后将该方法应用于铜单晶电极上的CO2电还原体系,准确地预测了平衡电位和电还原过电位。
长期以来,发展适用于金属表面催化体系的组合方法的重点是如何合理地分割离域体系。例如,Carter小组提出的嵌入簇模型,要求保持金属簇和金属表面的电子化学势相等。该方法尽管原理上严格,但在实际应用过程中的表现仍然差强人意。
然而,通过波恩-哈伯循环发现,直接影响表面反应能量学的是气相生成能和吸附键,这些因素具备显著的定域性。我们的前期研究结果表明,通过表面物种的生成能(即气相生成能和吸附键之和),可以方便地确定反应网络中决速步和决速态(ACS Catal. 2021, 11, 9333)。因此,尽管整个体系同时存在离域性和定域性,表面催化过程更关注定域的吸附键,其精度有望通过已有的组合方法高效地实现。这为发展适用于金属表面催化体系的组合方法提供了新的认识。此外,定域性的重要性,也意味着广泛应用的GGA泛函难以准确描述多相催化体系。

图1. (a)组合方法XYG3:GGA计算吸附能示意图。(b) Cu(111) 面采用的嵌入簇构型。(c)Cu(100)面所使用的嵌入簇构型。
如图1所示,我们通过XO-PBC,组合了周期性的GGA和高精度的双杂化泛函XYG3,实现了吸附能的精确高效计算。其中,周期性的GGA可以作为低层(PBC@L)准确地考虑离域性效应,而GGA描述定域性吸附键所产生的误差则可以在嵌入簇(Cluster@H)上由XYG3校正。
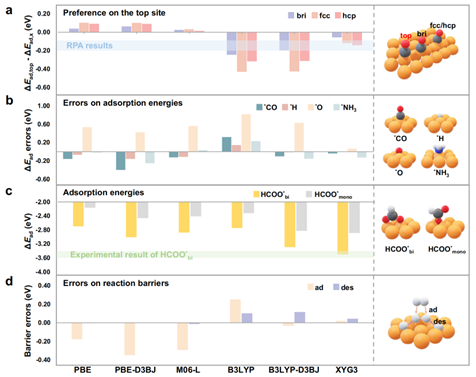
图2.方法测评。(a)最稳定的CO吸附位点;(b)分子吸附能误差;(c)HCOO*自由基吸附能;(d)H2的吸附和脱附能垒。meta-GGA、杂化泛函和双杂化泛函的计算均通过组合方法H:PBE-D3BJ计算。
图2通过一系列精确的实验值,测评对比了GGA泛函(PBE,PBE+D3BJ)、meta-GGA泛函(M06L)、杂化泛函(B3LYP,B3LYP+D3BJ)和双杂化泛函(XYG3)。测试集包括“CO Puzzle”难题、小分子和自由基吸附能、以及反应能垒。测试结果验证了XYG3:GGA的精度,相比于其他泛函精度优势凸显。
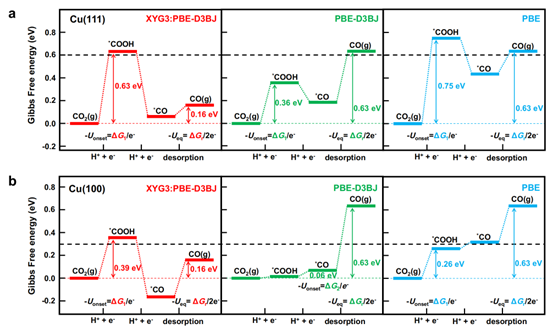
图3. XYG3:PBE-D3BJ方法应用于铜单晶电极上CO2电还原体系。虚线为实验过电位对应的反应自由能。
最后,我们将XYG3:PBE-D3BJ方法应用于铜单晶电极上的CO2电还原体系,准确地预言了平衡电位和电还原过电位(图3)。而GGA泛函的预测结果则存在较大的误差。PBE-D3BJ甚至预言了错误的电位限制步。
这一成果近期发表在Nature Communications上,文章的作者是复旦大学的陈征博士和刘章云博士,通讯作者是复旦大学徐昕教授。
Nat. Commun.2023, 14 (1), 936
https://doi.org/10.1038/s41467-023-36695-7